KOSIN UNIVERSITY COLLEGE OF MEDICINE
KOSIN UNIVERSITY COLLEGE OF MEDICINE
Articles
- Page Path
- HOME > Kosin Med J > Volume 34(1); 2019 > Article
-
Case Report
Lethal Cardiac Complications in a Long-Term Survivor of Spinal Muscular Atrophy Type 1 - Min-Jung Cho
-
Kosin Medical Journal 2019;34(1):47-51.
DOI: https://doi.org/10.7180/kmj.2019.34.1.47
Published online: June 30, 2019
Department of Pediatrics, Pusan National University Hospital, Biomedical Research Institute of Pusan National University Hospital, Busan, Korea.
- Corresponding Author: Min-Jung Cho, Department of Pediatrics, Pusan National University Hospital, Biomedical Research Institute of Pusan National University Hospital, 179, Gudeok-ro, Seo-gu, Busan 49241, Korea. Tel: +82-51-240-7800, Fax: +82-51-248-6205, mjchomd@gmail.com
• Received: August 23, 2017 • Revised: October 19, 2017 • Accepted: October 25, 2017
Copyright © 2019 Kosin University College of Medicine
Articles published in Kosin Medical Journal are open-access, distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/4.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
- 812 Views
- 1 Download
Abstract
- Spinal muscular atrophy (SMA) is a rare neuromuscular disease characterized by degeneration of the anterior horn cells of the spinal cord and motor nuclei in the lower brainstem, resulting in hypotonia, progressive proximal muscle weakness, paralysis, and progressive respiratory insufficiency. We report the case of a 6-year-old girl diagnosed with spinal muscular atrophy type 1 (Werdnig-Hoffman disease) who has been treated at home with non-invasive ventilation (assist-control mode with a back-up respiratory rate of 26 per minute). She presented with an atrioventricular block and atrial fibrillation, as well as paroxysmal fluctuation of blood pressure and heart rate indicating autonomic dysfunction. Although it is known that patients with spinal muscular atrophy type 1 do not generally demonstrate cardiac problems, it can be concluded based on findings in our case that long-term survivors with spinal muscular atrophy type 1 may develop cardiac rhythm disturbances. We therefore recommend that the possibility of cardiac complications and autonomic dysfunction should be borne in mind in the management of such patients.
- The patient was a 6-year-old girl with a long-standing diagnosis of SMA type 1. She had never been able to sit without support, and showed generalized muscle hypotonia with weakness. Diagnosis of SMA type 1 was confirmed at 2 months of age based on molecular genetic studies. At the age of approximately 8 months, she underwent placement of a gastrostomy feeding tube, and was treated at home with non-invasive ventilation (assist-control mode with a back-up respiratory rate of 26 per minute) due to swallowing dysfunction and progressive respiratory insufficiency. Her mother was a nurse, and she cared for her by herself without prescribed drugs or routine clinic follow-up.
- She was first referred to the cardiology department about the decreased heart rate that her mother had noticed. Her electrocardiogram showed a 2:1 high-grade second-degree atrioventricular block (Fig. 1). Her body weight was 18 kg. Pulse rate was approximately 50 beats per minute (bpm). Nonetheless, other vital signs were stable; 110/60 mmHg of blood pressure, 26 per minute of respiratory rate, 36.5℃ of body temperature, and 99% of percutaneous saturation. Her mother denied any relevant drugs including herbal medication or infection. Laboratory evaluation revealed a normal complete blood count, serum creatinine, blood urea nitrogen, and electrolyte profile. Echocardiography showed no structural or functional heart abnormalities. During continuous electrocardiographic monitoring, continuous 2:1 high-grade second-degree atrioventricular block improved to intermittent atrioventricular block pattern. As her vital sign was stable without other significant symptoms, she was discharged home. Twenty days after her first visit, she returned to the emergency department with complaints of excessive perspiration, episodes of cyanosis, and postprandial discomfort that had been observed at home over 5 days prior to presentation. At presentation, she showed a 2:1 high-grade second degree-atrioventricular block with a mean escape beat of 50 beats per minute, and her oxygen saturation was 99%. However, after admission, she demonstrated recurrent episodes of significant fluctuations in her heart rate up to 190 bpm, associated with fluctuation in her blood pressure (70/40 mmHg to 130/100 mmHg) and body temperature (36.3℃ to 38.5℃) in the absence of any inciting factors to account for such findings. Electrocardiographic monitoring documented several episodes of paroxysmal atrial fibrillation (Fig. 2). Five hours after presentation, abrupt cardiac arrest developed; she could not be recovered despite cardiopulmonary resuscitation.
CASE
- This case is a rare example of the occurrence of cardiac complications in a patient diagnosed with SMA type 1. Major complications observed in all forms of SMA are diffuse symmetrical proximal muscle weakness and progressive respiratory insufficiency. However, several reports have described cardiac complications like cardiac rhythm abnormalities and cardiomyopathy in SMA type 2 and type 3 patients. Elkohen et al.6 investigated 8 patients with SMA type 3 (Kugelberg-Welander disease); 2 of them, showed an atrioventricular block and a dilated cardiomyopathy. Furthermore, there is another case of Kugelberg-Welander disease who presented with syncope; the patient needed pacemaker implantation as showed a complete atrioventricular block without escape rhythm.7
- As opposed to cases with SMA type 2 or type 3, evidence of cardiac involvement in SMA type 1 is limited. Previous publications reporting cardiac involvements in patients with SMA type 1 have described either structural abnormalities such as atrial and ventricular septal defects8 or profound sinus bradycardia. A survey investigating 63 patients with SMA type 1 found that 24% of these patients presented with severe symptomatic sinus bradycardia, and four of them died because bradycardia progressed to a state of cardiac standstill.9 Our patient showed an atrioventricular block and atrial fibrillation, which, to date, have not been reported in patients with SMA type1. We assume that long-term survivors of SMA type 1 may demonstrate concomitant cardiac arrhythmias detected during their follow up.
- Another interesting finding noted in this case was fluctuation in blood pressure, body temperature, and heart rate demonstrated by the patinet. Hachiya et al. have reported similar autonomic dysfunction in a long-term survivor of SMA type 1.10 Their case described the presence of paroxysmal blood pressure and heart rate fluctuation in the patient. They observed high plasma concentrations of norepinephrine during phase of tachycardia, suggesting the role of autonomic instability in this condition, which was attributed to the involvement of the autonomic nervous system in childhood-onset motor neuron diseases.
- Although a majority of patients with SMA type 1 are known to die before 2 years of age as a consequence of respiratory insufficiency, the development of promising treatment strategies has changed the natural history and survival period observed in patients with SMA type 1.4 It is likely that a greater number of SMA type 1 patients with cardiac problems might be observed in the future. Our case suggests that the role of autonomic dysfunction and cardiac rhythm disturbance should be considered as another cause of morbidity and mortality associated with this disease, and these entities ought to be evaluated in long-term survivors of SMA type1.
DISCUSSION
- 1. Kolb SJ, Kissel JT. Spinal Muscular Atrophy. Neurol Clin 2015;33:831–846.ArticlePubMedPMC
- 2. Butchbach ME. Copy Number Variations in the Survival Motor Neuron Genes: Implications for Spinal Muscular Atrophy and Other Neurodegenerative Diseases. Front Mol Biosci 2016;3:7.ArticlePubMedPMC
- 3. Nicole S, Diaz CC, Frugier T, Melki J. Spinal muscular atrophy: recent advances and future prospects. Muscle Nerve 2002;26:4–13.ArticlePubMed
- 4. Oskoui M, Levy G, Garland CJ, Gray JM, O'Hagen J, De Vivo DC, et al. The changing natural history of spinal muscular atrophy type 1. Neurology 2007;69:1931–1936.ArticlePubMed
- 5. Chung BH, Wong VC, Ip P. Spinal muscular atrophy: survival pattern and functional status. Pediatrics 2004;114:e548–e553.ArticlePubMed
- 6. Elkohen M, Vaksmann G, Elkohen MR, Francart C, Foucher C, Rey C. [Cardiac involvement in Kugelberg-Welander disease. A prospective study of 8 cases]. Arch Mal Coeur Vaiss 1996;89:611–617.PubMed
- 7. Takahashi N, Shimada T, Ishibashi Y, Sugamori T, Hirano Y, Oyake N, et al. Cardiac involvement in Kugelberg-Welander disease: a case report and review. Am J Med Sci 2006;332:354–356.ArticlePubMed
- 8. Rudnik-Schöneborn S, Heller R, Berg C, Betzler C, Grimm T, Eggermann T, et al. Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J Med Genet 2008;45:635–638.ArticlePubMed
- 9. Bach JR. Medical considerations of long-term survival of Werdnig-Hoffmann disease. Am J Phys Med Rehabil 2007;86:349–355.ArticlePubMed
- 10. Hachiya Y, Arai H, Hayashi M, Kumada S, Furushima W, Ohtsuka E, et al. Autonomic dysfunction in cases of spinal muscular atrophy type 1 with long survival. Brain Dev 2005;27:574–578.ArticlePubMed
References
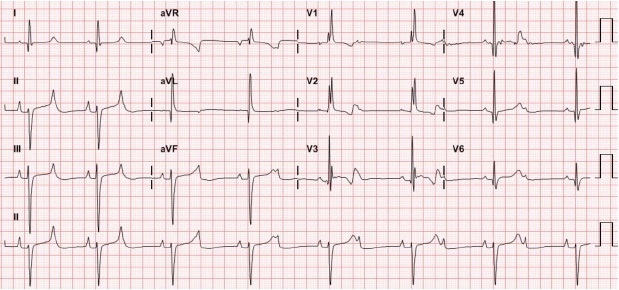
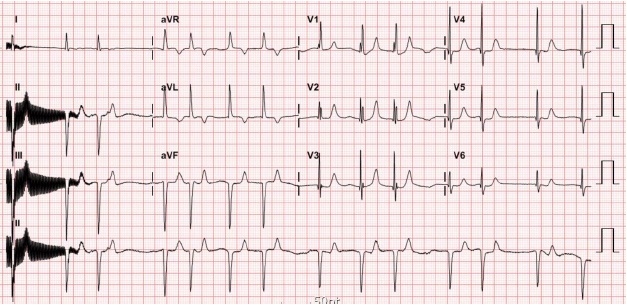
Figure & Data
References
Citations
Citations to this article as recorded by 
 PubReader
PubReader ePub Link
ePub Link Cite
Cite